Imaging & Sensing: NGS
Lessons learned from sequencing: a smart beginning for meaningful results
Massive sequencing, or next-generation sequencing, has opened the door to the fascinating world of genetics, and the secret on how to use this powerful tool most effectively hinges on the starting point: the sample
Lilian Martínez at Miltenyi Biotec
Next-generation sequencing (NGS) has developed exponentially over the past decade, unlocking vast research opportunities in genomics. High-throughput systems and powerful computational tools enable new applications from whole-genome analysis to single-cell resolution, driving discoveries that were once out of reach. With this enormous progress, critical questions arise regarding the selection of the NGS application that best suits the scientific aim, the type of sample required and the most optimal strategies for sample preparation to achieve reliable sequencing results.
The classic NGS workflow begins with sample preparation of the sample, followed by library preparation, in which the sample is converted into a form that can be sequenced, typically indexed DNA. After sequencing, the workflow finalises with data analysis (Figure 1).
It is often assumed that the more input material and deeper the sequencing, the more meaningful the data output will be. However, data quality depends not only on the input quantity but also on whether the sample is truly representative. As sequencing technologies have rapidly advanced, sample preparation methods have also evolved to meet the increasing demand for high-quality sample delivery, necessary to achieve successful sequencing results. Not all sequencing technologies provide standardised sample preparation protocols, leaving researchers to select the most suitable method on their own. While standard protocols may be available and widely used from specialists in the field, specific experimental requirements can demand alternative approaches. The decision about the sample preparation method should be made before library preparation and sequencing, ideally in consultation with the sequencing facility.
How to choose the most fitting NGS application
The first step in the scientific method is to ask a question. Clearly defining the scientific question is essential for choosing the appropriate NGS application and its corresponding sample preparation strategy. A well-structured plan prevents unnecessary pitfalls, such as poor sample quality or inappropriate sequencing depth, and increases the likelihood of generating meaningful data.
“Sample material is the starting point of the NGS workflow, and every step of its preparation, from handling complex tissues to preserving cell viability, directly shapes data quality”
Consulting literature, established protocols and bioinformatic expertise is essential for designing a robust NGS workflow. Reviewing how other researchers have applied NGS tools to similar questions can provide valuable guidance, and consulting a bioinformatician can help define the most suitable NGS approach, for example, whether a bulk approach is sufficient or if single-cell resolution might result in a more meaningful output. In bulk sequencing approaches, the heterogeneity of the sample can be a critical factor: the results for low-abundance cells may be diluted by information from all other populations in the sample. In such cases, isolating the populations of interest prior to sequencing can be beneficial to increase the sensitivity of the NGS application. On the other hand, single-cell sequencing captures data from individual cells, which is particularly useful in immunological studies that require the classification of cell subpopulations and link specific T-cell or B-cell receptor clonotypes to their gene expression profiles. Successful sequencing results depend on the sensitivity of the method, the number of input cells and adequate sequencing depth to achieve sufficient coverage.
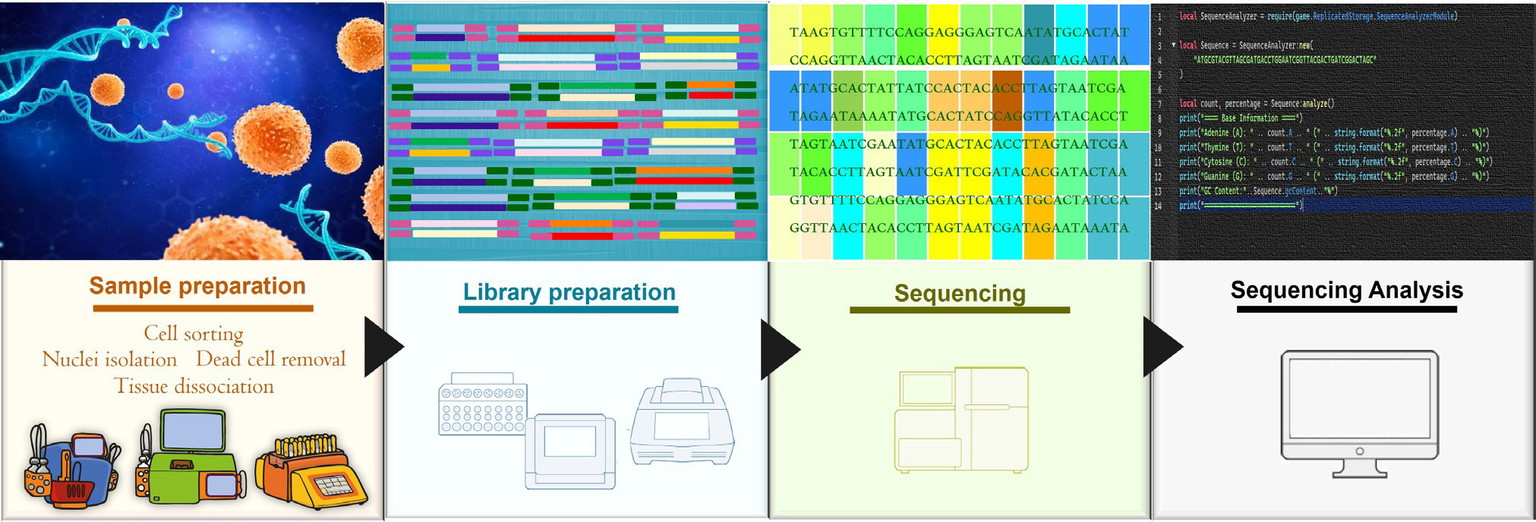
Figure 1: Classic NGS workflow. The standard NGS process begins with sample preparation, continues with library preparation and sequencing, and ends with data analysis
Preparing for success: optimising sample quality
Once the NGS approach is defined, optimising sample preparation is critical for successful results, as sample quality can significantly affect data reliability and interpretation. Strategies now exist to handle difficult samples, including those with low viability, rare cell types or poor-quality tissues. The following sections present some examples of such challenges and provide recommendations based on real-life cases from everyday laboratory work.
Managing low cell viability
For RNA-based NGS applications, particularly single-cell sequencing, several library preparation kit providers recommend a minimum total cell viability of 70%. In the case of samples with lower cell viability, removal of dead cells is recommended, as these can release RNA – known as ‘ambient RNA’ – which increases background noise and utilises sequencing reads.1 Various approaches for dead cell removal are available. Antibody-conjugated beads that bind to specific markers on dead cells have proven to be fast, cost-effective, scalable, gentle on cells and easy to use.2 Another approach is microfluidic-based cell sorting methods, which gently isolate viable cells with high purity while minimising cellular stress during the removal of dead cells and debris.3 Importantly, the chosen method for removing dead cells must fit the sample requirements of the NGS application, and remain compatible with downstream processes.
Strategies for rare cell sequencing
Cells that account for less than 3% of the total count are considered rare.4 Studying such rare cells can provide important insights into diseases such as cancer. However, their reliable and sensitive detection by NGS is challenging without prior isolation. Antibody-based cell sorting can be an effective strategy, but choosing the right antibodies is key. When no specific antibodies are available or the interaction of the rare population with other cells in the sample could be significant, it is possible to enter the NGS workflow with a higher total cell count. However, the chosen workflow must be able to accommodate the higher input material and generate sufficient sequencing depth to cover all cells in the sample.
Handling fragile cells
When working with cells that can be easily damaged or destroyed during processing, some workflow steps may need to be optimised. For example, minimising the centrifugation speed while extending centrifugation time, or using swing-bucket rotors, can minimise shear forces and allow gentle pellet formation. Special buffers with protein additives, such as bovine serum albumin, can further reduce cellular stress during sample processing. In some cases, alternative methods such as nuclei isolation may be a better alternative, achievable through bead-based separation or flow cytometry.5 Since sequencing quality depends directly on nuclei integrity, visual inspection of the isolated nuclei prior to library preparation is an important quality-control step and can be decisive for proceeding with the workflow.
Preparing tissue samples
In both clinical and research laboratories, tissue samples are often the starting material for NGS workflows. The optimal dissociation method depends on the properties of the tissue, such as its physical characteristics (eg, firm, mucous) and storage conditions (eg, fresh, frozen, fixed).6 Transport conditions are also critical: using appropriate storage and stabilisation media helps to avoid cell death during transfer from the collection site to the laboratory. Tumour tissues can be particularly challenging due to inherent cell decay and high levels of cell debris, making specialised storage media and optimised dissociation protocols essential.
Fixation is another important factor, as fixation protocols can directly affect the quality of the input material. In bulk NGS applications, especially RNA-based ones, the overall quality of the dissociated cells influences the quality of the total RNA. Single-cell sequencing, however, is even more susceptible to low-quality samples containing high proportion of dead cells and debris. Therefore, dissociation methods are often followed by removal of dead cells and targeted cell sorting, to maximise sample quality and achieve reliable sequencing results.
T-and B-cells: to sort or not to sort?
The decision depends on the experimental aim and the NGS application. Bulk applications specifically targeting the T-cell or B-cell receptors are highly sensitive and well suited for profiling the entire immune repertoire, as they allow a large amount of input for library preparation. Single-cell sequencing enables deeper insights, such as chain pairing and overlap with the transcriptomics profile, but it comes with higher costs.
Increasing input can improve single-cell workflows, but when resources are limited, pre-sorting to enrich target cells may be highly beneficial.7 The choice of the sorting strategy should be based on both the proportion of target cells in the starting material and the desired percentage in the final sorted material.
Conclusion
NGS has revolutionised genomics and offers numerous applications for diverse aspects of research. Sample material is the starting point of the NGS workflow, and every step of its preparation, from handling complex tissues to preserving cell viability, directly shapes data quality. Flow cytometry-based methods support NGS by enriching target populations, removing dead cells and debris, and protecting fragile cells. This precise handling of input material increases sensitivity and the ability to gain meaningful insights. With the right strategy, researchers can overcome the challenges of sample preparation and achieve reliable results that drive discovery.
References
- Yang S et al (2020), ‘Decontamination of ambient RNA in single-cell RNA-seq with DecontX’, Genome Biol, 5, 21(1), 57
- Fiorini MR et al (2025), ‘Ensemblex: an accuracy-weighted ensemble genetic demultiplexing framework for population-scale scRNAseq sample pooling’, Genome Biol, 3, 26(1), 191
- Staunstrup NH et al (2022), ‘Comparison of electrostatic and mechanical cell sorting with limited starting material’, Cytometry A, 101(4), 298-310
- Wang X et al (2024), ‘MarsGT: Multi-omics analysis for rare population inference using single-cell graph transformer’, Nat Commun, 15, 338
- Visit: protocols.io/view/nuclei-isolation-and-clean-upusing-gentlemacs-and-3byl4w2k2vo5/v1
- Alasfoor S et al (2024), ‘Protocol for isolating immune cells from human gastric muscularis propria for single-cell analysis’, STAR Protoc, vol 5(3), 103258a
- Laghmouchi A et al (2017), ‘Long-term in vitro persistence of magnetic properties after magnetic bead-based cell separation of T cells’, Scand J Immunol, 92(3), e12924

Lilian Martínez leads the Next Generation Sequencing Core Facility at Miltenyi Biotec. Her team provides complete counselling from experimental design to sequencing data generation, while working closely with the NGS bioinformaticians. She holds a degree in Biological Chemistry from the University of San Carlos, Guatemala, with a major in Clinical Laboratory Management, and a Master’s and PhD in Genetics from the Institute of Human Genetics at University of Cologne, Germany. Lilian has 15 years of experience in the public and private health sector, forensic research and IVD-development in biotechnology companies such as QIAGEN, Hilden.